Development News Brief
On this page
Get Galaxy

new: $ hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist
upgrade: $ hg pull -u -r e6444e7a1685
## BLAST+ Migration
The tool set NCBI BLAST+ has moved from the Galaxy distribution to the Galaxy Main Tool Shed.
Migration scripts will run upon Galaxy’s first launch (after updating to this release) that will automatically handle installing BLAST (and blastxml) from the Tool Shed.
## Reference Genome rsync Server
If you would like to obtain the same reference genome builds and indexes as available on the public Galaxy Main instance, these can retrieved from the rsync server at:
datacache.g2.bx.psu.eduFor example, to download the complete directory for the ‘phiX’ genome:
$ rsync -avzP rsync://datacache.g2.bx.psu.edu/indexes/phiX .Genomes are organized in directories by the dbkey. If you are not sure of the dbkey, check your datasets. The dbkey is what is populated into the “database” attribute for a dataset. Read more about how this fits into data integration or [setting up native genome indexes](/admin/NGS Local Setup/).
## More Updates to Output and Error Handling
As reported in the July 20th, 2012 News Brief, several changes have been made to the underlying code that determines run result state from tool exit codes and output. There are now additional enhancements to applying regular expressions and exit code checks. Read more…
## Tools
Admin/Config/Tool Dependencies
- Enhancements
- Tophat2 wrapper enhancements:
- Include fusions output. Read more about what this is in the Tophat2 Manual’s section Fusion mapping options:
- Bowtie2 wrapper enhancements:
- Output sorted BAM from Bowtie2 by default
- One benefit is that BAM results can be used as input to Cufflinks without an intermediate sorting step.
- NOTE: If you are using an older version of Bowtie or uploading your own results, sorting is still required before running Cufflinks, whether in SAM or BAM format.
- Tophat2 wrapper enhancements:
- New
- Galaxy RNA-seq Analysis Exercise on Main
- Walks through sample protocol step-by-step using paired-end data, initial read QC through CuffDiff analysis
- Includes iGenomes sourced reference annotation GTF, an answer key, and bonus resources
- Galaxy RNA-seq Analysis Exercise on Main
## User Interface (UI)
- Enhancements
- Does not use
enable_tracksorenable_pagesoptions anymore; visualizations and pages are enabled for all instances.
- Does not use
- New
- Addition of interactive phylogenetic tree visualization.
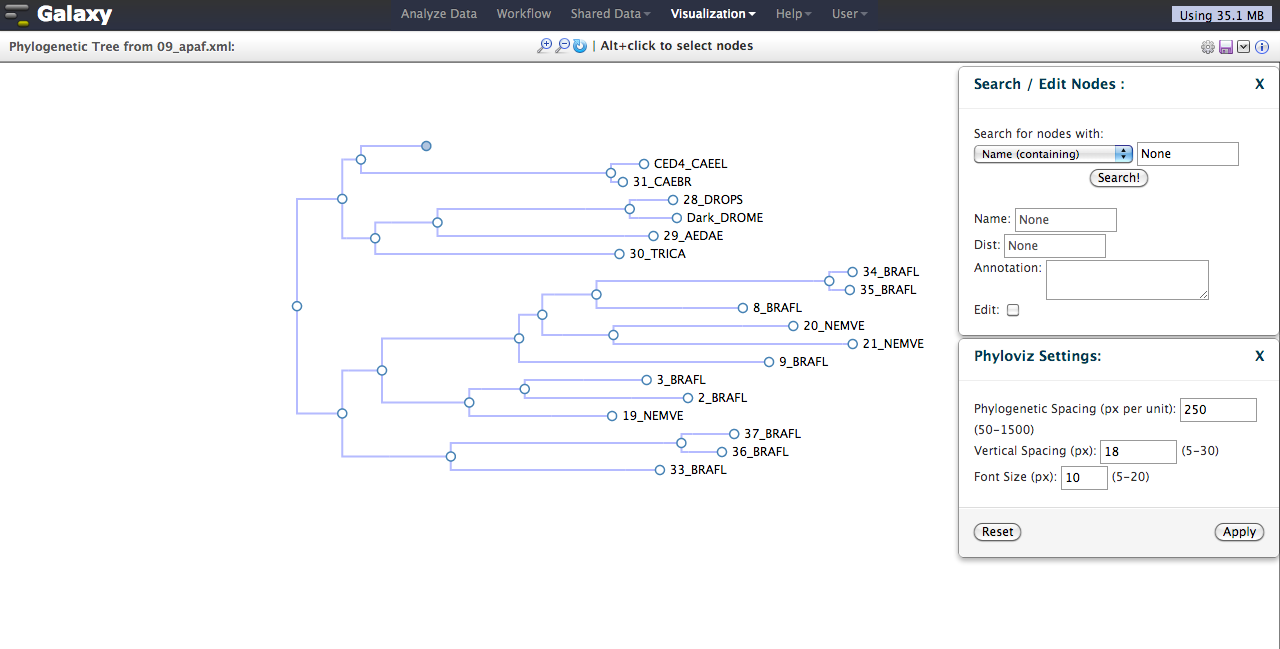
## Galaxy Track Browser (GTB)
- New
- Enhancements
- Enable visualization of bedGraph datasets
- Server-side code for coverage histograms
- Add feature search to Trackster ; typing in location box will search tracks in visualization for features that start with entered text. Works with GFF, GTF, and BED datasets. Fixes https://bitbucket.org/galaxy/galaxy-central/issue/611
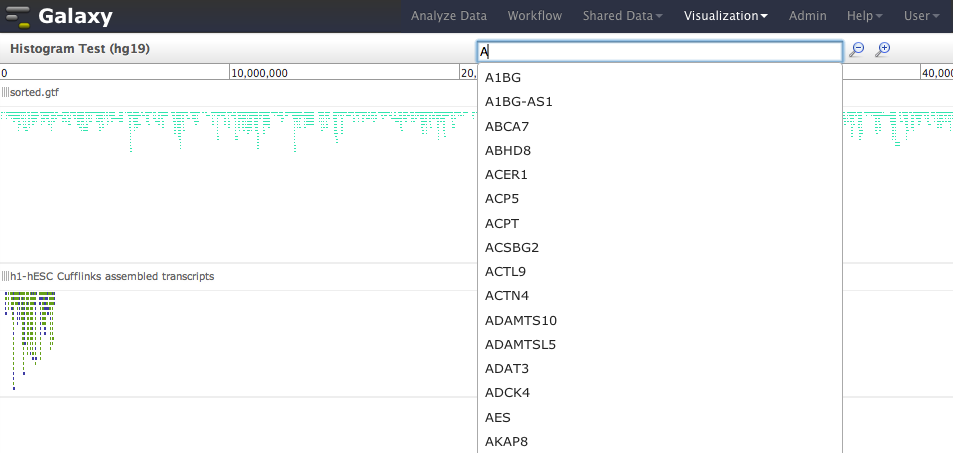
## Source
- New
- A native job runner for the Condor DRM, submitted by Jaime Frey
scripts/db_shell.pyis an interactive shell for working with the Galaxy model, contributed by John Chilton
- Enhancements
- New enhancement that adds a security mechanism to the lwr job runner, contributed by John Chilton
- If using
run.sh,'$GALAXY_UNIVERSE_CONFIG_DIR'can be set to a directory containing partial config files, which will be merged in to a single universe_wsgi.ini - Binary datatype sniffing has been moved to the datatype classes and removed from the upload tool thanks to a contribution by John Chilton. Local modifications to the upload tool may conflict and should no longer be necessary. See pull request 59 for examples.
- Removed
contrib/multiprocess.shhas been removed. If you’re using multiple Galaxy processes, set'GALAXY_RUN_ALL=1'in your environment and start/stop as usual with:
$ sh run.sh --daemon/sh run.sh --stop-daemon## Workflows
- New
- New parent tag copying for multiple workflow run output histories, contributed by Brad Langhorst. See pull request 54
- Enhancements
- Workflow API changes to support parameter execution and workflow creation, in collaboration with Richard Park. See pull request 55
- Additional API changes, contributed by John Chilton. See pull request 62
## Tool Shed
- Fixes
- Several miscellaneous fixes for using a SQLite database with a local tool shed.
- It is no longer possible to change the name of a repository in the tool shed at during the time when the repository is first being cloned.
- Setting metadata on tool shed repository changeset revisions has been re-engineered, resulting in the resolution of several critical issues. Repository metadata is now generated for appropriate changeset revisions, and repository contents are now displayed correctly for each changeset revision for which metadata can be generated.
- Error messages have been improved for tool shed repositories that include invalid tools. Clicking on an invalid tool in your repository should provide you the information needed to correct the tool.
- Tool section labels are now handled correctly in the tool panel (attempting to remove them or change their location in the tool panel used to be problematic).
- Entries defined in the
tool_shed_conf.xmlfile no longer require a trailing ’/’ in the defined urls. For example, the following entry used to be necessary:
Now the above entry still works, but the following entry is also ok (notice the ’/’ character after the port number in the url is no longer necessary):
<tool_shed name="My local tool shed" url="http://localhost:9009"/>- Enhancements
- The Tool Shed is now running Mercurial version 2.2.3.
- The Freebayes repository was updated in the Main Galaxy Tool Shed to Revision: 7:d3bf1e86b243, so make sure to get the updates if you have installed it into your local Galaxy instance. For details about getting updates to your installed repositories, see this section of the Galaxy tool shed wiki Getting updates for tool shed repositories installed in a local Galaxy instance
- The implementation for importing proprietary datatype class modules included in a tool shed repository now supports class module files whose name conflicts with a Python standard library module name. For example, if a proprietary datatype class module is named
xml.py, it will now be correctly imported even though the name conflicts with the Python standard library’s xml module. - The repository tip is now displayed in a column that is separate from the repository’s installable changeset revisions in the tool shed.
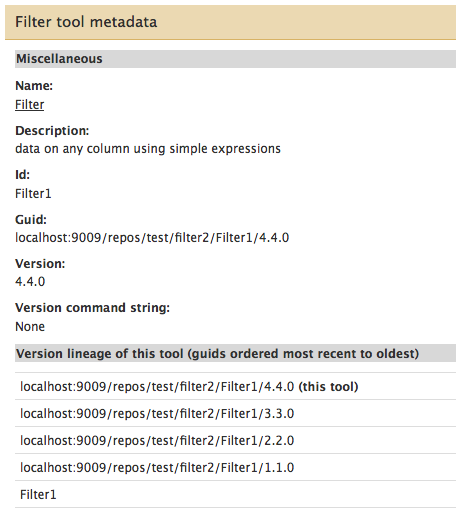
- Additional information has been added to the “Tool metadata” page, which is displayed when you choose the “View tool metadata” option from a tool’s pop-up menu. This menu is available in 2 locations: when viewing the repository in a tool shed, and when viewing a repository (which includes tools) that has been installed into a local Galaxy instance. The additional information added to the Tool metadata page includes:
- information about tool dependencies if they have been defined in the repository
- requirements defined in the tool config
<requirements>tag set - The tool’s version lineage information, an example of which is shown here for a sample tool named Filter
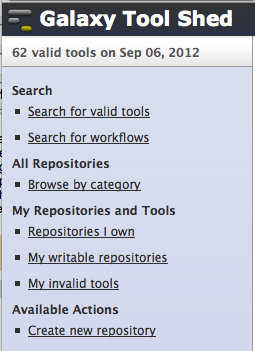
- In addition to browsing repositories that you own, you can now also browse repositories for which you have been granted “write” permission. Here is a snapshot of the new Galaxy tool shed menu.
## Bug Fixes
## Announcements
News, August and September 2012 Galaxy Updates

Highlights
- Aug
- GCC2012 & GCC2013: slides and video
- 29 new papers
- Tool Shed Contributions
- Sept
Swiss Galaxy Day

The 1st Swiss Galaxy Workshop will be held October 3-4 in Bern, and is aimed at Galaxy administrators and users alike. We also welcome participants who are using other workflow management systems, and tool developers who are looking for such systems to offer their tools to a wider audience.
We would like to discuss the status of the Galaxy project, new developments, interface to other systems, extensions and best practice in reproducible research.
The workshop is part of the SyBIT Tech Day series.
### Who's Hiring
Got a Galaxy-related opening? Send it to outreach@galaxyproject.org and we’ll put it in the Galaxy News feed and include it the next Galaxy Update and News Brief.
## About Galaxy
The Galaxy Team is a part of BX at Penn State, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State, and Emory University.
Join us at Twitter @galaxyproject or just read our tweets Galaxy on Twitter